
This is a proof-of-concept post based on the TensorFlow library developed by Google. The idea is simple: using a publicly available microbiome dataset, I wish to apply deep-learning algorithms for potential diagnostic platform development.
Copyright Declaration
Except for the image above this declaration, and unless otherwise stated, the author asserts his copyright over this file and all files written by him containing links to this copyright declaration under the terms of the copyright laws in force in the country you are reading this work in.
This work is copyright © Ali A. Faruqi 2016. All rights reserved.
Introduction
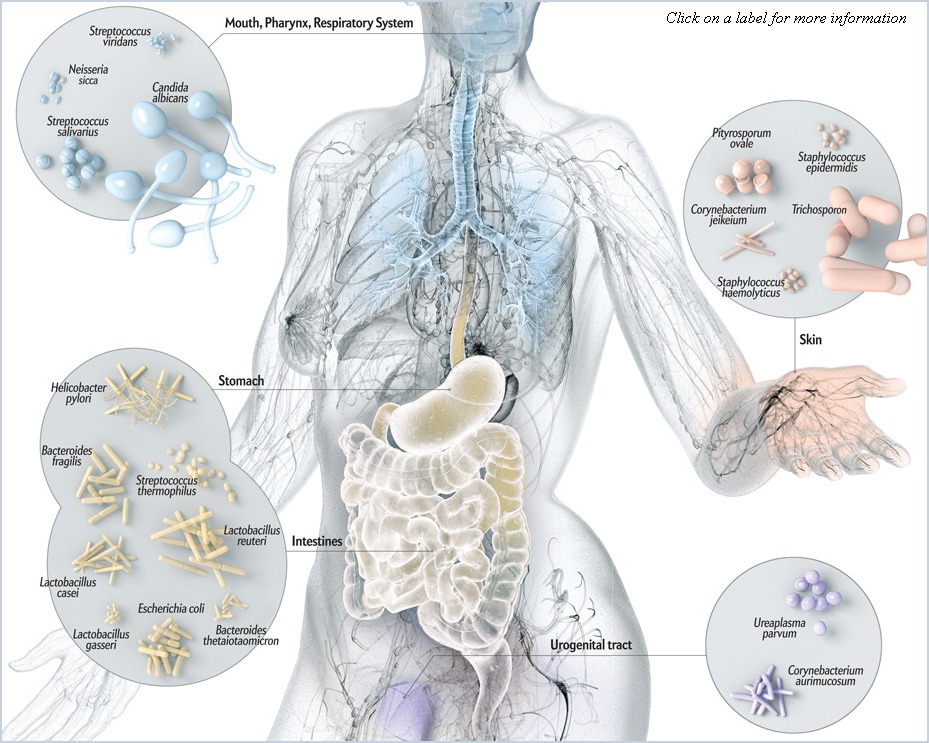
What is the Human Microbiome?
A succinct answer to the question asked above, as obtained from the Human Microbiome Project website, is as follows:
- The Human Microbiome is the collection of all the microorganisms living in association with the human body.
- These communities consist of a variety of microorganisms including eukaryotes, archaea, bacteria and viruses.
- Bacteria in an average human body number ten times more than human cells, for a total of about 1000 more genes than are present in the human genome.
Many of these microbes are essentially helpful for human beings and can help in the following ways:
- Producing vitamins that we do not have the genes to make.
- Breaking down our food to extract nutrients we need to survive.
- Teaching our immune systems how to recognize dangerous invaders.
- Producing helpful anti-inflammatory compounds that fight off other disease-causing microbes.
The business of microbiome
According to a new market research report titled: “Human Microbiome Market by Indication (Obesity, Cancer), Application (Therapeutic, Diagnostic), Product (Prebiotics, Food, Drugs), Product Research (Instruments, Consumables), Technology Research (High Throughput, Omics Technology) - Global Forecast to 2023”, published by MarketsandMarkets, the microbiome market is expected to reach USD 658 Million by 2023 from USD 294 Million in 2019 growing at a CAGR of 22.3% during the forecast period (2019-2023).
There is great potential for growth in and capitalization of the microbiome market. Large companies such as DuPont, JNJ and Merck along with small startups such as Second Genome, Whole Biome, Phylagen and ubiome, to name a few, are catering to this market.
Microbiome-based diagnostics
The composition of our microbiomes has been shown to be correlated with numerous disease states by growing amount of scientific literature. The human microbiome, therefore, has an important role in the diagnosis and early detection of various diseases.
Microbiome data can be leveraged to achieve one of the goals of systems biology in making medicine predictive, preventive, personalized and participatory. This is known as P4 medicine.
In the following sections, I have developed a simple deep-learning model using the microbiome data to predict the HIV status of individuals based on a simple Softmax Regression model.
Technical Details
Data collection
For the purpose of demonstration, I will focus on a single study carried out by Lozupone et al. The study by Lozupone et al showed an association of the gut microbiome with HIV-1 infection. Using this knowledge, I obtained the dataset for this study, which is made publicly available by the Qiita microbiome data storage platform.
The dataset obtained from Qiita for Lozupone’s study had HIV-1 status information available for 51 individuals. 30 individuals are HIV-1 positive and 21 individuals are HIV-1 negative.
Model building
Given the limited human and computational resources I had access to i.e., myself and my laptop, I built a fairly simplistic softmax-based regression model.
A microbiome dataset consists of a count matrix in which the bacteria characterized by Next Generation Sequencing (NGS) are the rows and the samples from which the bacterial count information is obtained are columns. Additionally, there is meta-data associated with the samples. On a technical note, the bacterial count here refers to OTUs.
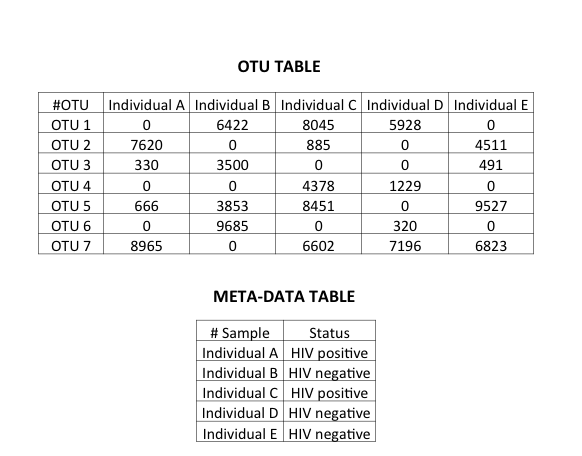
It is beyond the scope of this post to go into the definition of OTUs. Suffice to say, I am treating bacteria/OTUs as input features in the model. On a related note, OTU tables are sparse matrices.
The response variable is the disease state. In this particular dataset, it is the status of HIV-1, which is either positive or negative. The dataset had information about 267 bacteria/OTUs i.e., features. The training set had 29 samples and testing set had 22 samples.
Model evaluation
Using the input data for Lozupone’s study, I wrote a simple Python script that ran softmax-regression. The script and further technical specs are on my GitHub repository called TFMicrobiome.
Even though the dataset was small and the model simple, I ended up getting an accuracy of 91%! This demonstrates that even the most simplistic of deep-learning system is very powerful in dealing with real-life noisy data and has the potential to be developed into a diagnostic platform.
Future Directions
Moving forward, there are three broad areas where effort needs to be put in.
Other biological datasets
I have made use of only a small microbiome dataset. From a statistical perspective, the sample size is small and there is a need for larger datasets to train the models.
Additionally, and from a biological point-of-view, microbiome is part of the picture. At a molecular level, there are other high-throughput datasets available such as that of the genome, transcriptome, proteome and metabolome. By integrating the various ‘omics’ datasets for a given individual, we can develop a truly systems level understanding of various biological diseases and achieve the objectives of P4 medicine.
Other deep learning algorithms
As a simple POC, I made use of the simplistic softmax regression model. It is not the most powerful method out there. Moving forward, I’d like to see some more sophisticated algorithms being employed such as convolutional neural networks and recurrant neural networks, to name a few.
Feature selection in deep-learning
One of the aims of medicine is to make inference. That is, given the diagnosis, how can one know etiology of the disease and develop appropriate intervention and prevention strategies. In the context of deep-learning for microbiome, the aim is not just to make a prediction about disease. It is also about feature selection. We’d like to know which features i.e., microbes, are important in causing or preventing the disease. This will help in the development of therapeutics. The challenge, therefore, is to develop a deep-learning system that can, apart from making accurate prediction about diseases, can also provide information of about the microbes, genes, proteins and metabolites that impact the health status.