
This is a proof-of-concept post based on the TensorFlow library developed by Google. To the best of my knowledge, it is the first ever attempt to apply Convolutional Neural Networks on microbiome data.
Copyright Declaration
Except for the image above this declaration, and unless otherwise stated, the author asserts his copyright over this file and all files written by him containing links to this copyright declaration under the terms of the copyright laws in force in the country you are reading this work in.
This work is copyright © Ali A. Faruqi 2016. All rights reserved.
1. Introduction
In a previous post, I discussed about the microbiome, its market capitalization and its potential to be used in diagnosis of diseases. I further demonstrated how even a simple deep-learning algorithm, as implemented in TensorFlow, can be applied on a given microbiome dataset and provide not-so-bad accuracy.
Being fairly new to the field of neural networks and being completely self-taught, I decided to move ahead and apply a more sophisticated model on the same dataset. I chose convolutional neural networks. Convolutional neural networks are a type of artificial neural network that are inspired from the biological connectivity of neurons of the animal visual cortex.
There is extensive literature on convolutional neural networks (CNN) and it is the beyond the scope of this post to do an extensive survey on CNNs. In this post, I will provide a description of applying CNNs to microbiome data as a proof-of-concept exercise. This is the first time, to the best of my knowledge, that CNNs have been applied to microbiome data. So, without further ado, let’s dive right into CNNs.
2. Technical Details
2A. Reading data
The input file of microbiome data comes as an OTU table. The individual (sample) on which microbiome data is obtained is actually a vector of OTUs. In the Lozupone et al dataset, we have information on 267 OTUs. Our input for CNN, therefore, is a 267x1 vector for each sample.
2B. Steps of CNN
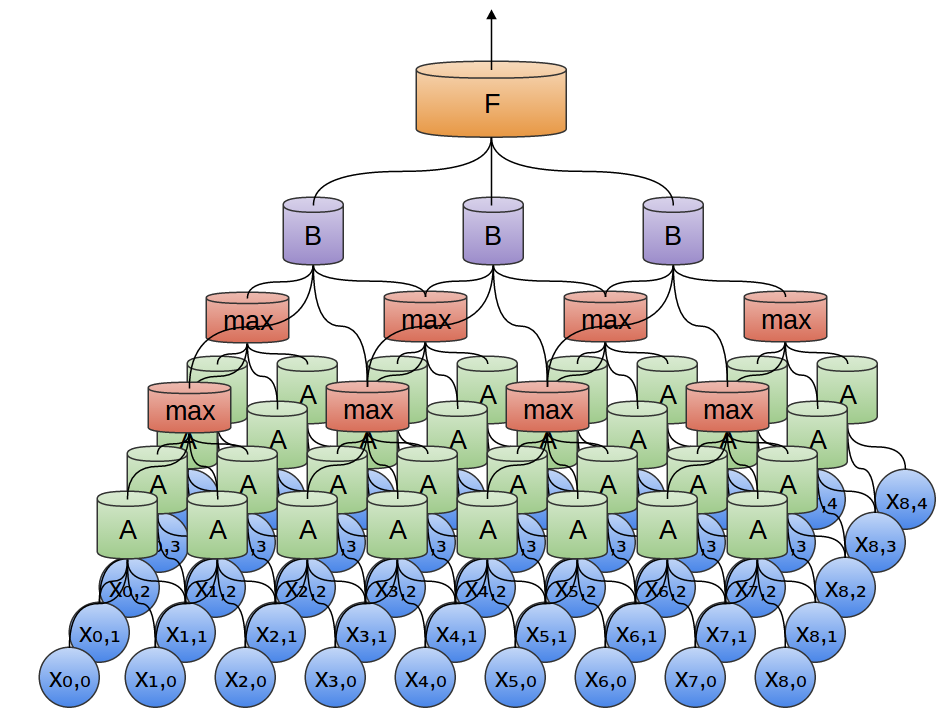
Convolution layers and max pooling are the main elements of CNNs. In the simple Python script I’ve written called cnn.py, the method proceeds as shown in the following figure:

To elaborate the figure a bit more:
- The OTU table is input for the first convolutional layer followed by max-pooling.
- The output of first max-pooling is input to second convolutional layer followed by max-pooling.
- The output of second max-pooling is input to densely connected layer.
- Dropout is performed on the output of densely connected layer.
- The output of dropout is finally passed to the readout layer, which performs simple softmax regression.
2C. Input reshaping
In traditional image classification datasets, such as MNIST, one would like to reshape the flattened image pixel vector back to the original 2d image array.
tf.nn.conv2d method in TensorFlow library, which computes a 2-D convolution given 4-D input and filter tensors can be used for this purpose. Unlike the MNIST dataset, in which the in_height and in_width of the input tensor can be set to 28x28, in the microbiome dataset, the in_height and in_width are set to 1 and 267 respectively, where 267 is the number of OTUs (features) in the dataset.
3. Results
Even though CNN model is more sophisticated than basic softmax regression, we get a much lower accuracy (of about 0.772) for our dataset using CNN. I have tried playing around with different variables and parameters. Following are some of the variables I have tried to manipulate.
- Train-to-test split ratio: The highest accuracy I got was with train to test ratio of 0.55.
- Max-pooling: Whether one does max-pooling or not, it did not seem to have an impact on accuracy with the current microbiome dataset.
- Dropout: Like max-pooling, doing or not doing dropout does not seem to be impacting the accuracy.
- Gradient descent optimizers: AdadeltaOptimizer with learning_rate = 0.1, rho = 0.95 and epsilon = 1e-02 seems to be giving accuracy of 0.77. I have also tried AdamOptimizer and it seems to be giving comparable results. I haven’t tried other optimizers.
- Stride for conv2d and maxpool: The highest accuracy is when the stride of the sliding window is 1 for both conv2d and maxpool at the start of the script i.e., the input to the first convolutional layer.
- Changing activation functions: Changing the activation functions from relu to relu6, elu, sigmoid, tanh, softplus and softsign didn’t change the accuracy.
- Bias variable initialization: I changed the value of bias variable initialization from 0.1 to 1 to 10 to 100 but did not see increase in accuracy.
Few variables that I have not explored so far include:
- Changing the number of convolutional layers
- Changing the output channel values of convolutional layers
- Randomizing the training and test datasets
4. Discussion
One reason for the relatively poor performance of convolutional neural networks over softmax regression, maybe, has to do with the type of input data. CNNs are great for tasks such as image classification because they can take into account the spatial structure of the data. In image classification, for example, input pixels which are spatially closer together would have higher correlation than pixels far apart.
In the microbiome dataset, however, there is no corresponding spatially local correlation that CNNs can exploit by enforcing a local connectivity pattern. One way to develop an analog for spatially local correlations in microbiome data is to utilize the phylogenetic relationship information between OTUs. That is, OTUs that are evolutionarily closer can have higher correlation than OTUs that are evolutionarily distant. This is something I can explore in future posts.
Summing up, even though CNN has not done a better job compared to basic softmax regression, there is potential for further fine tuning this model. Larger microbiome datasets can be used for this purpose, evolutionary information can be taken into account and further parameter tuning can be done in order to improve the predictive power of the model.